معرفی بیماریهای ژنتیکی -در حال حاضر میلیونها نفر در سراسر جهان به بیماریهای ژنتیکی مبتلا هستند و وراثت و سابقه خانوادگی مهمترین فاکتور برای پیشبینی خطر ابتلا به بیماریهای ژنتیکی است. همچنین تاکنون تحقیقات متعددی برای شناسایی و درمان این بیماریها انجام شده است.
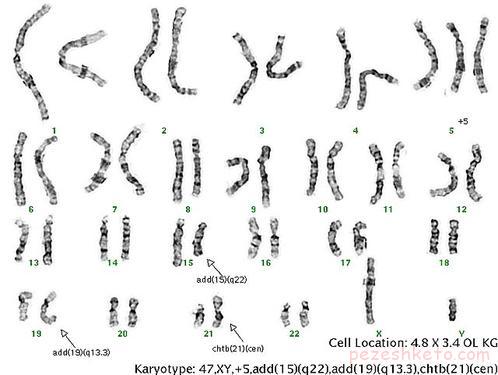
یک اختلال ژنتیکی از طریق ایجاد ناهنجاریهایی در فاکتورها و عناصر تعیین کننده صفات و ویژگیهای ژنتیکی فرد شامل مولکولهای دیانای یا ژنوم (توالی ژنها) ظاهر میشود و این ناهنجاریها به چهار گروه جهش تکژنی، چندژنی، تغییرات کروموزومی و میتوکندریال تقسیمبندی میشوند.
همچنین بر محققان مشخص است که برخی نژادها مستعد ابتلا به اختلالهای ژنتیکی خاص هستند به گونهای که ابتلای آنان به این بیماریها از پیش تعیین شده است. به طور مثال ساکنان مناطق مدیترانهای اروپا بیشتر مستعد ابتلا به نوعی کمخونی ژنتیکی هستند و برخی اختلالهای ژنتیکی همچون کمخونی داسی شکل، حاصل واکنش بدن برای مقابله با محرکهای محیط زیست است.
در واقع در بیماران مبتلا به کمخونی داسی شکل، نوعی جهش ژنتیکی که موجب میشود گلبولهای قرمز خون تغییر شکل دهند در به حداقل رسیدن خطر آلوده شدن فرد به انگل مالاریا کمک میکند.
در این گزارش به بررسی شایعترین و کشندهترین انواع بیماریهای ژنتیکی پرداختهایم که به شرح زیر است:
سندرم داون
سندرم داون یک ناهنجاری کروموزومی شایع است که از تکثیر اضافی ژنهای موجود در کروموزوم 21 ناشی میشود. از هر 800 تا 1000 نوزاد یک نوزاد به این سندرم مبتلا میشود که میتوان در آزمایشهای پیش از زایمان آن را تشخیص داد. همچنین برخی از مشخصههای ظاهری این بیماری همچون تغییر در شکل صورت، کاهش انعطاف عضلات و نارسایی سیستم گوارشی و قلب بلافاصله پس از تولد ظاهر میشوند. احتمال ابتلا به این سندرم در تمام نوزادان وجود دارد اما در نوزادانی که مادرانشان در سن بالا آنها را باردار شدهاند این احتمال بیشتر است.
سندرم ایکس شکننده
سندرم ایکس شکننده با شایعترین نوع عقبافتادگی ذهنی و اختلال ارثی تاخیر در رشد مرتبط است. این اختلال رشدی و مشکلات ذهنی میتواند از نوع خفیف تا شدید متفاوت باشد و گاهی اوقات نیز با اوتیسم همراه میشود. آمار نشان میدهد که از هر 1500 مرد و 2500 زن یک نفر به این سندرم مبتلا میشود. به طور معمول مردان یک کروموزوم X و زنان از این کروموزوم دو عدد دارند اما در مبتلایان به سندرم ایکس شکننده، بخشی از کروموزوم X از هم جدا میشود. این قسمت شکنندگی روی کروموزوم X اگر در نقاط دیگر همین کوروموزوم تکرار شود، بیماری وخیمتر میشود.
اختلال ارثی لختگی خون
فرآیند لخته شدن خون یکی از پدیدههای زیست شیمیایی پیچیده در بدن است و انواع مختلفی از اختلال ارثی لختگی خون وجود دارند. این مشکل میتواند به خونریزیهای بسیار و تشکیل لختههای خونی غیرعادی در تمام نقاط بدن و عمدتا در رگها بینجامد و مشکلات و ناراحتیهای جدی برای فرد مبتلا بوجود آورد.
دیستروفی ماهیچهای
بسیاری از موارد دیستروفی ماهیچهای که با ضعیف شدن ماهیچهها شناخته میشود جزو اختلالات وراثتی به حساب میآیند. این اختلال به دو نوع دیستروفی ماهیچهای «دوشن» و «بکر» تقسیم میشود.
در نوع اول این بیماری علائم آن پیش از سن شش سالگی ظاهر میشود که شامل خستگی، ضعف ماهیچه و عقبافتادگی ذهنی است. همچنین میتواند با علائم دیگری همچون مشکلات قلبی، تنفسی و بد شکلی سینه و پشت ظاهر شود. به گفته پزشکان، پسران بیشتر این بیماری را به ارث میبرند. نوع دوم دیستروفی ماهیچهای نیز علائمی مشابه با نوع اول دارد با این تفاوت که با سرعت و شدت کمتری ظاهر میشود.
تالاسمی
بیماریهای تالاسمی گروهی از اختلالهای ارثی خونی را شامل میشوند که به موجب آن گلبولهای قرمز به درستی هموگلوبین تولید نمیکنند. در اثر ابتلا به این بیماری کمخونی ظاهر میشود که شایعترین علائم آن شامل خستگی، شکستگی استخوان، درد استخوانی، تنگی نفس و بزرگ شدن طحال است. همچنین بروز عفونتها در مبتلایان به این بیماری شایع است.
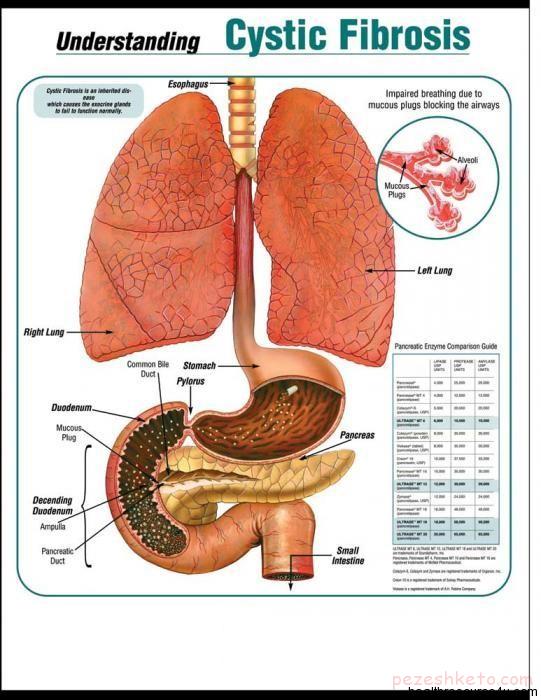
فیبروزسیستیک
فیبروزسیستیک شایعترین و مرگبارترین بیماری ژنتیکی است که تنها در آمریکا نزدیک به 30 هزار نفر به آن مبتلا هستند. این بیماری باعث میشود بدن مخاطهای غلیظ و چسبندهای تولید کند که سبب مسدود شدن ریهها شده و فرد را دچار عفونت میکند و بر عملکرد پانکراس نیز تاثیر میگذارد. تنفس در این بیماران به سختی انجام شده و با متوقف شدن فعالیت آنزیمهای گوارشی از جذب مواد مغذی جلوگیری میشود.
بیماری تای ساکس
این بیماری موجب میشود تا افراد مبتلا به آن در هر سنی جان خود را از دست بدهند، هرچند معمولا پنج سالگی سن بروز این بیماری و دلیل آن، فقدان آنزیم «هگزوزآمینیداز» است. بیماری تای ساکس موجب تخریب سیستم عصبی و مغز میشود که غیر از کنترل کردن علائم بیماری، راهحل درمانی برای آن وجود ندارد.
الکلیسم
اعتیاد به الکل که یک بیماری در نظر گرفته میشود از طریق ترکیبهای ژنتیکی به نسل بعد منتقل میشود. در صورتی که یکی از والدین یا هر دوی آنان به مصرف الکل اعتیاد داشته باشند، احتمال آنکه فرزندان آنها به مصرف الکل اعتیاد پیدا کنند نیز افزایش مییابد که در بسیاری از موارد میتواند سرطانزا و کشنده باشد.
سرطان سینه و روده
از هر 9 زن، احتمال دارد یک نفر به سرطان سینه مبتلا شود. وجود نسخههای ژنی BRCA1 و BRCA2 که از والدین به ارث میرسد، زنان را در معرض ابتلا به سرطان سینه و تخمدان قرار میدهد. با این حال تمام موارد ابتلا به سرطانهای سینه از دلایل ژنتیکی ناشی نمیشوند. همچنین نقش سابقه خانوادگی در ابتلا به سرطان روده به این معنی است که میتواند آن را به عنوان یک بیماری ارثی در نظر گرفت.
کمخونی داسی شکل
وجود حالت تک ژنی برای سلول داسی شکل به خودی خود عامل بیماری نیست و این ژن فرد را در برابر ابتلا به بیماری مالاریا محافظت میکند اما دریافت این ژن از هر دوی والدین موجب میشود گلبولهای قرمز حالت غیرعادی پیدا کرده و داسی شکل شوند. این سلولهای داسی شکل به مویرگهای کوچک میچسبند و به تخریب مفاصل و اعضای بدن منجر میشود.
چاقی
چاقی بیماری پیچیدهای است که بر اثر محرکهای زیستمحیطی و ژنتیکی ناشی میشود. فرزندان والدین چاق، بیشتر در معرض خطر ابتلا به چاقی هستند.
بیماری قلبی
بسیاری از انواع بیماریهای قلبی از طریق فاکتورهای ژنی به نسل بعد منتقل میشوند. به طور خاص سندرم «بروگادا» به عنوان بیماری ژنتیکی قابل درمان است. در صورتی که یکی از اعضای خانواده به این بیماری که با ضربان نامرتب قلب همراه است مبتلا شود، احتمال ابتلای افراد دیگر به این بیماری افزایش مییابد.
هموفیلی
این اختلال شامل خونریزی ناشی از فقدان فاکتورهای ژنتیکی لخته کننده خون است که از پدر یا مادر یا هر دوی آنان به ارث میرسد. جایگزین کردن این فاکتور، راه کنترل اختلالهای متعدد خونریزی است. همچنین از آزمایشهای خاصی برای تعیین و شناسایی فاکتور لخته شدن خون که در بدن فرد مبتلا به هموفیلی وجود ندارد، استفاده میشود.
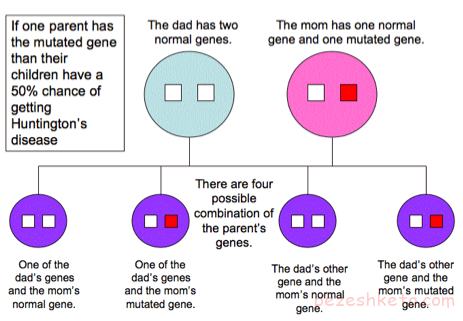
بیماری هانتینگتون
این بیماری ژنتیکی به آرامی و با تاثیرگذاری بر عملکرد شناختی و وضعیت نورولوژی فرد مبتلا، منجر به مرگ وی میشود.
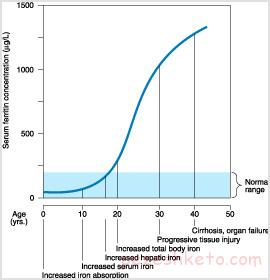
هموکروماتوز
بیماری «هموکروماتوز» بر توانایی بدن در تنظیم جذب آهن تاثیر میگذارد که عدم درمان آن میتواند آسیبهای جدی به اعضای بدن وارد کند. خارج ساختن خون سرشار از آهن از بدن، سریعترین و امنترین شیوه برای درمان این بیماری است.
کوررنگی
بررسیها نشان میدهد حدود 10 میلیون مرد آمریکایی نمیتوانند رنگ قرمز را از سبز تشخیص دهند، هرچند این اختلال در زنان آمریکایی کمتر از 600 هزار نفر است. دلیل ابتلای فرد به این عارضه این است که ژنهای دریافت کننده نور قرمز و سبز در کروموزوم X کنار یکدیگر قرار میگیرند. مردان، تنها یک کروموزوم X دارند که از مادر خود به ارث میبرند. در حالی که زنان دو کروموزوم X دارند و یک ژن طبیعی روی کروموزوم X دیگر عملکرد ژن معیوب را متوازن میسازد و به همین دلیل مردان بیشتر در معرض ابتلا به این اختلال هستند.
خشن بودن
محققان معتقدند ژنی که افراد را در معرض انجام رفتارهای خشن قرار میدهد، شناسایی کردهاند. به گفته محققان، رفتارهای تهاجمی در پسران بیش از رفتارهای دیگر احتمال دارد که به ارث برده شوند. هرچند در زنانی که دست به سرقت میزنند، نقش ژنها بیشتر است.
آکنه
بررسیها نشان میدهد پسرانی که در سن مدرسه به بیماری پوستی آکنه مبتلا میشوند در سوابق پزشکی خانوادگیشان نیز این بیماری وجود دارد.
عدم تحمل لاکتوز
ابتدا تصور میشد بیمیلی مردم چین به خوردن شیر عامل فرهنگی دارد اما محققان در دهه 1960 میلادی عدم تحمل لاکتوز را در میان ساکنان آسیا، آفریقا و اروپای جنوبی شناسایی کردند که عامل ژنتیکی و ریشه در نژاد آنها دارد.
طاسی
طاسی معمولا در مردان شایع است. هر چند ژنها در بروز این بیماری نقش دارند، مادران تنها علت بروز آن در فرزند به حساب نمیآیند. طاسی معمولا بر اثر وجود ناهنجاری در چندین ژن منتقل شده از پدر و مادر یا هر دوی آنان اتفاق میافتد.
نوروفیبروماتوز
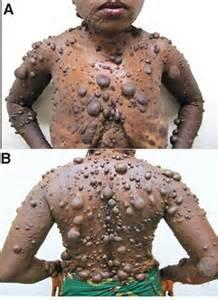
«نوروفیبروماتوز» گروهی از اختلالهای ژنتیکی هستند که از طریق رشد تومورها در سراسر سیستم عصبی بدن شناخته میشوند و سه نوع اصلی آن نوروفیبروماتوز 1، نوروفیبروماتوز 2 و اسکوآنوماتوز هستند که نوع اول آن شایعتر است. در هر سه نوع از این بیماری، ژن معیوب از تولید پروتئینهای ضروری جلوگیری میکند که منجر به تقسیم سلولی غیرکنترل شده میشود.
همچنین از آنجا که این تومورها میتوانند در هر نقطه از سیستم عصبی ظاهر شوند، تاثیرات بیماری نوروفیبروماتوز به طور قابل توجهی بین بیماران متفاوت خواهد بود. این بیماری معمولا کشنده نیست اما میتواند مشکلات جدی را همچون کم شنوایی، سرطان، تغییر شکل استخوان یا مشکلات شناختی را برای فرد به وجود آورد.
سندرم آنجلمن
این سندرم اختلالی است که با ویژگیهایی همچون بروز مشکل در سیستم عصبی یا تاخیر در تکامل شناخته میشود و علائم این بیماری معمولا در اولین ماههای تولد ظاهر میشوند. افراد مبتلا به این بیماری معمولا برای خوردن غذا دچار مشکل میشوند. حدود سن دو سالگی فرد دچار تشنج شده و قادر به صحبت کردن نیست.
با بزرگتر شدنِ کودک مبتلا به سندرم آنجلمن، مشکلاتی در مهارتهای حرکتی، بیش فعالی، اختلال خواب و کوچک شدن سر مشاهده میشوند. این بیماری ارثی کشنده نیست هر چند درمانی برای آن کشف نشده است. همچنین افراد مبتلا به این سندرم، بیاختیار میخندند و همیشه لبخند میزنند که دلیل آن مشخص نیست.
بیماری پلیکیستیک کلیه
این بیماری اختلالی ژنتیکی است که موجب میشود چندین کیست غیرسرطانی در کلیههای بیمار تشکیل شوند. در حالی که این تومورها خوش خیم هستند، میتوانند مشکلاتی همچون سنگ کلیه، نارسایی کلیوی و فشارخون بالا را در فرد ایجاد کنند. اغلب مبتلایان به این بیماری از آن آگاهی ندارند و زمانی به پزشک مراجعه میکنند که وزن کلیههایشان به 14-9 کیلوگرم رسیده باشد.
منبع:isna.ir
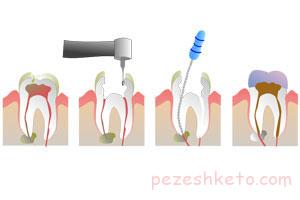 مراحل عصب کشی دندان
مراحل عصب کشی دندان