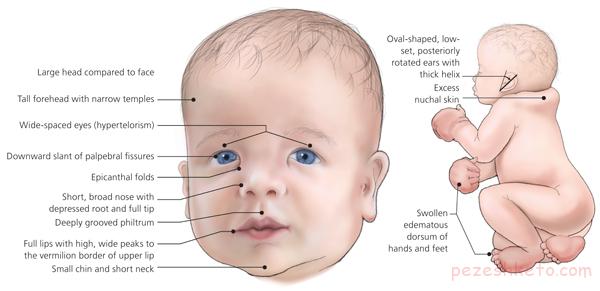
سندرم نونان یك اختلال ژنتیكی است كه موجب اختلال در تكامل و رشد طبیعی قسمتهای مختلف بدن می شودد. خصوصیات ظاهری بارز، قامت كوتاه، نقص قلبی و سایر ناهنجاریهای جسمی و عقب ماندگی ذهنی از مشخصات این سندرم است. تخمین زده می شود كه یک نفر از هر هزار تا 2500 تولد به این سندرم مبتلا باشند. سندرم نونان زمانی عارض می شود كه كودك یك كپی از ژن مبتلا را از والدینش دریافت می دارد. چنانچه هیچگونه تاریخچه ایی از بروز این سندرم در خانواده وجود نداشته باشد و كودك به این سندرم مبتلا باشد علت بروز این رویداد موتاسیون خود بخودی در كودك است. هیچ درمانی برای سندرم نونان وجود ندارد و درمان بر مدیریت علائم بیماری و عوارض مرتبط با آن متمركز است.
 علت سندرم نونان چیست :
علت سندرم نونان چیست :
علت سندرم نونان موتاسیون یكی از دو ژن مسئول برای تولید یك پروتئین خاص است كه نقش مهمی در تكامل قلب، سلولهای خونی، استخوانها و سایر بافتها ایفا می كند. این ژن جهش یافته ممكن است از یكی از والدینی كه حامل ژن معیوب است به ارث برسد و حتی ممكن است یك جهش جدید و تازه در كودكی كه هیچ زمینه ژنتیكی از بیماری نداشته رخ دهد. دو ژن شناخته شده وجود دارد كه ممكن است جهش یافته و سبب بروز سندرم نونان گردد.
– ژن :PTPN-11 در حدود نیمی از افراد مبتلا به سندرم نونان به علت جهش این ژن به این عارضه مبتلا می گردند. اسم این ژن “پروتئین تیروزین فسفاتاز غیر رسپتوری نوع یازده” است . این ژن به بدن دستور می داد كه چگونه پروتئین SHR2″ تیروزین فسفاتاز غیر رسپتوری نوع 11 ” را بسازد. SHR2 به سلولهای بدن كمك می كند تا بدانند وظیفه شان چیست و چه موقع باید تقسیم شوند. این امر در بدن فرآیند بسیار مهمی است كه در تكامل قلب، سلولهای خونی ، استخوانها و سایر بافتها قبل از تولد جنین نقش مهمی را بازی می كند.
اما دربسیاری از افراد مبتلا به سندرم نونان، ژن PTPN-11 بدرستی كار نمی كند. عقیده بر این است كه جهش ژنتیكی سبب می شود پروتئین SHR2 در تمام زمان روشن باشد و این در حالی است كه در حالت عادی این پروتئین در برابر واكنش به سیگنال هایی كه از سایر پروتئین ها دریافت می دارد باید خاموش و یا غیر فعال گردد. در نتیجه سیستمی كه در بدن كنترل عملكرد سلولها از قبیل رشد و تقسیم طبیعی را كنترل می كند بدرستی عمل نكرده و متعاقب آن نقص ها و ناهنجاری های جسمی رخ می دهد.
– ژن : KRAS در حدود 5 تا 10 درصد افراد مبتلا به علت جهش در این ژن به این عارضه مبتلا می گردند. این ژن مسئول ساخت پروتئینی در بدن است به نام K-RAS كه مسئول تنظیم تقسیمات سلولی است. این پروتئین سیگنال هایی را به سلولهای مغزی می فرستد و به آنها نحوه تقسیم و چگونگی رشد آنها را بیان می دارد. اینكه در چه زمانی باید چه عمل خاصی را انجام دهند. زمانی كه جهش در ژن KRAS رخ می دهد، پروتئین K-RAS به فعالیت خود ادامه می دهد حال آنكه نیاز است در برخی موارد خاموش و یا غیر فعال گردد. نتیجه این امر، رشد و تقسیم متوالی و بی پایان سلول ها و اختلال در روند رشد نرمال فرد و بلوغ بافتهای مختلف بدن می گردد. بروز این حالت قبل از تولد نوزاد بوده و كودك در هنگام تولد با نشانه های ظاهری و ناهنجاری های آشكار متولد می گردد.
علائم سندرم نونان چیست
– فقدان شنوایی: التهاب مزمن گوش میانی اغلب مشكلاتی را برای كودكان مبتلا به سندرم نونان را به بار می آورد. فقدان شنوایی در یک سوم افراد مبتلا به سندرم نونان مشاهده می گردد.
– مشكلات پوستی: این عارضه در افراد گوناگون متفاوت است اما بروز این سندرم تاثیر زیادی بر بافت پوست می گذارد كه پوست تمایل به كلفت شدن دارد بخصوص در نواحی كه اسكار جراحی وجود داشته باشد. افراد مبتلا به سندرم نونان اغلب مو های مجعد و ضخیمی دارند و یا حتی ممكن است موهای كم پشت داشته باشند.
– مشكلات ادراری تناسلی: بسیاری از افراد بخصوص مردان مبتلا به سندرم نونان دارای مشكلاتی در سیستم ادراری تناسلی هستند. مشكلات كلیوی محدود بوده و اغلب عده قلیلی از افراد مبتلا به این سندرم را درگیر خود می كند. ممكن است فرآیند بلوغ هم در دختران و هم در پسران به تاخیر بیفتد اما اغلب دختران باروری نرمال خواهند داشت اما این امر در مردان بعلت عدم نزول بیضه ها ممكن نرمال نباشد .
مشكلات لنفاوی: سندرم نونان سبب بروز مشكلاتی در سیستم لنفاوی می گردد بروز این حالت ممكن است قبل یا بعد تولد و یا بطور كانونی و یا پراكنده و وسیع خود را نشان دهد. بیشترین مشكل رایج تجمع مایع در پشت دستها و بالای پاهاست كه اغلب بطور خودبخودی در دوران كودكی رفع می گردد.
مشكلات عصبی: بروز علائم عصبی شیوع كمتر ی دارد اما در صورت بروز منجر به هیپوتونی در ماهیچه ها و باز شدن بیش از حد مفاصل می گردد. افراد مبتلا به سندرم نونان بندرت ممكن است دچار تشنج گردند.
– خونریزی: اغلب افراد مبتلا به سندرم نونان تاریخچه ایی از كبودی های غیر عادی و یا خونریزی دارند. اغلب اوقات مشكلات خونریزی تشخیص داده نشده تا زمانی كه فرد در معرض جراحی قرار گرفته و هموراژی را تجربه می كند.
– مشكلات چشمی: تقریبا تمام افراد مبتلا به سندرم نونان ناهنجاریهایی در چشم و پلك خود دارند. تفاوت در اندازه و شكل چشم ها از علائم این سندرم است. اغلب افراد مبتلا دارای عنبیه آبی كمرنگ و یا سبز هستند. افراد مبتلا به سندرم نونان همچنین دچار مشكلاتی در رابطه با ماهیچه های چشمی، مشكلات انكساری (دوربینی– نزدیك بینی – آستیگماتیسم) و مشكلات در اعصاب چشمی می باشند.
– ناتوانی هایی یادگیری: در حدود یک چهارم از افراد مبتلا به سندرم نونان دچار مشكلات یادگیری هستند و برخی نیز نیاز به آموزشهای خاص دارند. اما برخی از آنها قادر به كسب تحصیلات دانشگاهی می باشند
– بافتهای عضلانی – اسكلتی: شكل غیر معمول قفسه سینه به همراه استرنوم فرو رفته در 90 تا 95 درصد كودكان مشاهده می شود. اسكلیوز و كیفوز و گردن كوتاه و پوست اضافه نیز در این افراد شایع است.
– مشكلات قلبی: بین 50 تا 80 درصد كودكان مبتلا به سندرم نونان مشكلات مادرزادی قلب دارند. پزشك متخصص قلب كودكان ، اولین فردی كه كودك مبتلا به سندرم نونان را معاینه می كند .
تشخیص سندرم نونان
هیچگونه تست تشخیصی برای سندرم نونان وجود ندارد اما این سندرم بواسطه مالفورماسیون های ظاهری و جسمی قابل تشخیص خواهد بود. هر كودكی كه در آن احتمال وجود سندرم نونان وجود داشته باشد باید به منظور بررسی نقائص قلبی آزمایشات كامل قلبی صورت گیرد. تست های خونی به منظور بررسی كمبود فاكتور 11 و سایر فاكتور های انعقادی انجام می شود. گذاشتن نتایج تمامی تست ها در كنار هم ممكن است در تشخیص و اثبات این سندرم موثر باشد.
درمان سندرم نونان
درمان علائم و عوارضی كه در سندرم نونان رخ می دهد بسته به نوع و شدت آن دارد.
درمان قلب: داروهای خاصی ممكن است دربهبود برخی مشكلات قلبی مفید باشد اگر دریچه های قلبی درگیر باشند ممكن است نیاز به جراحی باشد و نیاز است كه قلب بطور دوره ایی مورد ارزیابی قرار گیرد.
درمان كاهش رشد: بسیاری از نوزادان مبتلا به سندرم نونان با سرعت نرمال رشد نمی كنند. كودكان مبتلا باید هر شش تا 12 ماه برای اطمینان از رشد كافی، مورد ارزیابی قرار گیرند. كودكان مبتلا ممكن است مشكلاتی در خوردن نیز داشته باشند و با داشتن این مشكلات قادر به تغذیه كافی نباشند. پزشك ممكن این نیاز را احساس كند كه عملكرد تیروئید و رشد استخوانی و سطح هورمونی كودك را مورد ارزیابی قرار دهد. اگر سطح هورمون رشد پایین باشد، هورمون تراپی ممكن است در درمان مفید واقع شود.
ناتوانی در یادگیری: طیف وسیعی از مسائل ذهنی و رفتاری در ارتباط با مبتلایان به سندرم نونان وجود دارد. خوشبختانه بسیاری از روش های موفقیت آمیز مهارتی وجود دارد كه برای كودكان با علائم خفیف مفید خواهد بود
درمان مشكلات عصبی: اگر كودك علائم و نشانه های عصبی از خود بروز می دهد پزشك ممكن است به ارزیابی كامل عصبی بپردازد كه شامل انواع آزمایشات تصویر برداری برای بررسی مغز می باشد. اگر كودك تشنج كند از داروهای ضد تشنج برای كودك تجویز شود.
درمان مشكلات ادراری تناسلی: اگر بیضه كودك پسر نزول نكرده باشد مممكن است تجویز HCG و یا جراحی لازم باشد. فرآیند درمان بهتر است قبل از ورود كودك به مدرسه صورت پذیرد. از دیگر مشكلات مردان این است كه ممكن است بیضه آنها عملكرد مناسب خود نداشته باشد، بنابراین جایگزینی تستوسترون یك گزینه درمانی محسوب می شود.
درمان مشكلات شنوایی: زمانی كه كودك نوزاد است باید ارزیابی های شنوایی صورت گیرد. این تست ها باید هر ساله تكرار گردد. اگر مشكل عفونت گوش باشد درمان سریع می تواند از فقدان شنوایی پیشگیری نماید. اگر در برخی موارد نیز شنوایی رخ دهد استفاده از سمعك مفید خواهد بود.
پیشگیری:
اگر تاریخچه ایی از وجود سندرم نونان در خانواده وجود داشته باشد مشاوره با پزشك و متخصص ژنتیك بخصوص زمانیكه تصمیم به بارداری وجود دارد مفید خواهد بود. با این حال به علت اینكه بسیاری از موارد بیماری ناشی از جهش خودبخودی است بنابراین راه پیشگیری وجود ندارد.
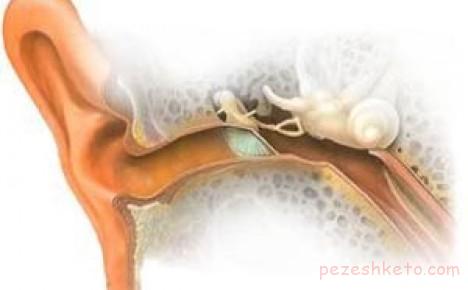
 به اين تومور وستيبولار شوانوما هم گفته مي شود چون منشاء اوليه تومور از عصب وستبرلار است.
به اين تومور وستيبولار شوانوما هم گفته مي شود چون منشاء اوليه تومور از عصب وستبرلار است.